使用生物信息学工具去撕下重复序列的面具(1)
在日常的生信分析中,我常常会碰到对重复序列的处理。对于重复序列知道是什么东西,也知道分析中需要避免它的影响。但实际上还是缺乏一些系统上的认识,感谢最近发的一遍nature genetics的review paper,帮我把这段知识补充完整。这里也分章节同时分享给大家,与大家一起学习。
摘要
首先好习惯先看看摘要:
许多物种的大部分基因组来自转座因子(TE)。此外,通过各种自我复制机制,TE在大多数物种的基因组中继续增殖。
TE已经提供了许多调控,转录和蛋白质创新,并且还与疾病有关。然而,尽管已经证实了它们的影响,许多基因组研究仍然排除它们,因为它们的重复性导致各种分析复杂性。幸运的是,正在开发的越来越多的方法和软件工具来满足它们。本综述介绍了与TE有关的生信工具,并重点介绍了进行TE全面基因组分析的一些挑战和目前所遇到的困境。
一贯经典nature genetics review的风格,简单的介绍,影响,现状,总结。没有一句拖泥带水,简单明了。
背景
1948年Barbara McClintock通过玉米开创性工作发现,转座子(TEs),已在所有植物和动物中被发现,以及各种原核物种。TE通常是根据中间底物繁殖插入(RNA或DNA)分为两类,并根据各种结构特征进一步分为家族和亚家族。 TEs负责主要的基因组扩增,TE衍生的序列构成了大多数真核基因组的大部分,包括大约一半人类基因组和高达95%的一些植物基因组。
大多数TE已经积累了突变和阳离子转变事件,使它们不再合适转换。此外,不同的物种已经进化出各种抑制机制,包括TE启动子甲基化,以防止进一步的转置事件。但是,在大多数基因组中,有一些
家族保持活跃并且可以产生新的插入,称为多态插入。例如,LINE-1家族仍在人类中活跃,并且还对许多其他家庭的动员事件负责(I类Alu元素和sVA元件)。据估计,对于LINE-1,每95个新生儿中有1个新种系插入频率,每21个新生儿中有1个Alu插入。这些LINE介导的插入反过来,它可以破坏整合位点的基因,已经有124个这样的插入与人类疾病相关。在人类中,许多TE家族在其他物种中仍然活跃,例如大多数植物,果蝇和小鼠的基因组中的长末端重复序列(LTR)家族以及植物和非哺乳动物物种中的DNA转座子。TE插入也可以在体细胞中发生并且已经在植物,秀丽隐杆线虫和哺乳动物基因组中观察到
。在人类中
已经有L1,Alu和SVA元素的体细胞插入,可以在神经细胞和癌细胞中观察到。
然后功能部分blabla的介绍一大段,忽略不写了。
尽管TE在正常功能和疾病产生中具有这些重要功能,但TE通常被忽略或
在基因组研究中“掩盖”,因为它们的重复性使得分析具有挑战性,特别是使用短读序列技术。例如,由于TE的reads通常是模糊比对的(比对到不同的序列中),这些模糊比对可能会被排除在下游分析之外。此外,使用短读取的组装的基因组通常难以正确放置TE,并导致下游注释不完整。尽管如此,我们正处于TE检测和分析技术进步的时代。实际上,降低成本导致了大规模测序项目的爆炸式增长,其中可以研究TE多态性及其影响。此外,还出现了新的长读取测序技术,这些技术降低了TE检测和基因组组装的复杂性。利用这些进步,正在开发多种方法和软件工具,以促进将TE纳入基因组研究。
这一段还是讲的相当好的,二三代技术的优劣势,简单的展望。
在本综述中,研究者提供了生物信息学工具的综合指南,这些工具是为检测和分析TE而开发的(图1)。首先,介绍各种可用的分类工具和数据库。接下来,专注于基因组序列中TE的注释工具,这些工具依赖于从头和目标方法。之后,我们描述了用于多态TE插入检测的主要策略,并探讨了一些关键的例子。我们还介绍了正在开发的新方法,以表征和预测TE的功能影响。最后,我们提供了许多标准工具,可以定制以在执行基因组分析时考虑TE,并总结分析TE时当前的一些挑战和目前的不足。

将文章的下面的内容铺垫好,接下来将讲述关于TE的4大部分。
TE分类和存储库
TE分为两大类,进一步分为族和子家族。关于TE的信息被编目为三种类型的存储库:以TE为中心,以基因组为中心和以多态性为中心。以TE为中心的存储库收集有关与每个TE家族相关的共有序列的信息,以基因组为中心的存储库对参考基因组中的所有单个TE实例进行编码,以及以多态为中心的存储库包含与该物种的注释参考基因组不同的个体中的插入。
以TE为中心
这些数据库专注于TE本身,包含每个家庭和亚家族的共识序列。它们用于分类目的,基因组中TE的注释,以及需要TE参考序列的各种其他生物信息学工具。 RepBase Update是真核基因组中最受欢迎的TE共识库,旨在包含每个TE家族的共识序列或代表性实例43。 RepBase Update将TE分为三组:DNA转座子,LTR反转录转座子和非LTR反转录转座子。 Dfam是一个更新的真核生物TE中心数据库,其中TE家族更正式定义,并通过隐马尔可夫模型收集为多序列比对。
Dfam还促进了TE个体的注释与运用于已知TE家族相关但已累积突变并远离共有序列的实例。 RepBaseUpdate和Dfam都与RepeatMasker一起使用,一种通过对与数据库中存在的序列同源的序列进行全基因组搜索来识别重复序列的工具用于注释人类基因组和大多数其他真核基因组基因组。
以TE数据库为中心,搜寻不同种类的TE就类似于,平时我们通过NR或者NT数据库寻找同源序列那样。通过与数据库的比较,将你的TE进行分类。
以基因组为中心
以基因组为中心的数据库编目在参考基因组中注释的各个TE。在基因组内和TE家族中显示其多样性,以基因组为中心的TE目录通过提供精确的TE序列来允许更准确的TE查询。现在人类,植物,真菌等都有相应的以基因组为中心的TE数据库。
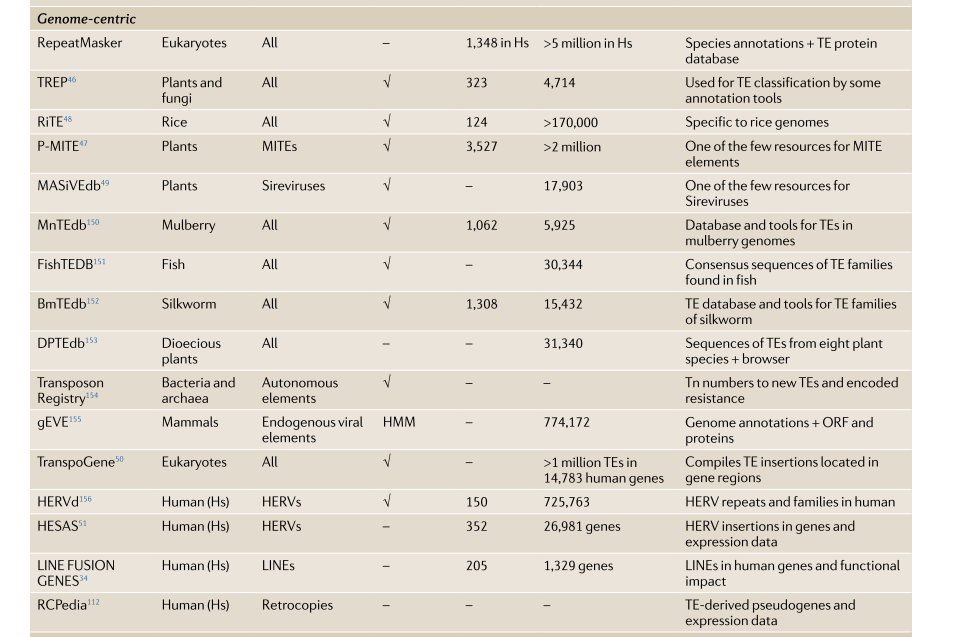 1.PNG-111.4kB
1.PNG-111.4kB
多态性为中心
在这些专用数据库中报告了在个体中检测到但在参考基因组中不存在的种系和体细胞多态性插入。随着对更多个体进行测序并发现新的插入,可以确定这些插入的种群频率。此外,这些数据库提供了更大的单个TE的混池来探索TE多样性。多态存储库还可以帮助将TE与不同的表型相关联,并且这些数据库中的一些还报告插入的假定功能影响。这些数据库都是特定于宿主的,并且通常是在大型重新测序项目的背景下创建的,这些项目旨在描述特定物种的多样性。同上人类,植物,果蝇都有自己相应的数据库:
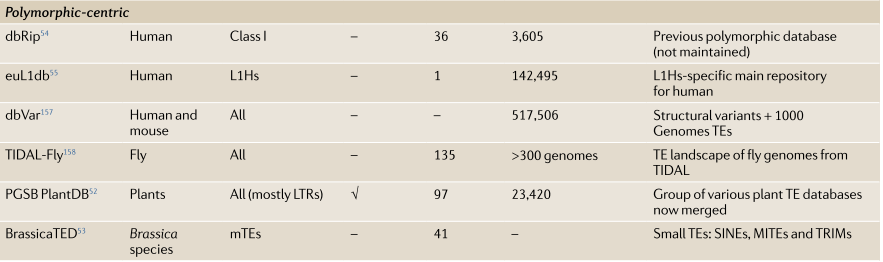 2.PNG-40kB
2.PNG-40kB
目前面临的挑战
TE数据库的两个方面仍然不是最理想的。首先,物种特异性储存库对于解释生物体内TE的序列多样性至关重要,但数据库之间存在一些重叠,如果将资源库与共享宿主或TE类型合并将有利于避免需要多个查询并增加凝聚性。其次,目前我们需要一个专门用于人类基因组中整合了不同方面TE多态性发现的综合资源。


